고성능 컴퓨팅(HPC) 기반 코로나19 연구에 대한 고든벨(Gordon Bell) 특별상 최종 후보는 대규모 언어 모델(LLM)에 유전체학, 역학, 단백질 공학에 대한 통찰력을 제공할 수 있는 새로운 언어인 유전자 염기서열을 훈련시켰습니다.
10월에 발표된 이 획기적인 연구는 아르곤 국립연구소(Argonne National Laboratory), NVIDIA, 시카고 대학(University of Chicago) 등의 20명 이상의 학계와 기업 연구원의 협업으로 이루어졌는데요.
연구팀은 LLM을 훈련하여 코로나19의 배후에 있는 바이러스인 SARS-CoV-2의 유전 변이를 추적하고 주요 변이 바이러스(VOC)를 예측했습니다. 지금까지 생물학에 적용된 대부분의 LLM은 소분자 또는 단백질의 데이터 세트에 대해 훈련을 받았지만, 이 프로젝트는 DNA와 RNA의 가장 작은 단위인 원시 뉴클레오타이드 서열에 대해 훈련을 받은 최초의 모델 중 하나입니다.
이 프로젝트를 주도한 아르곤 연구소의 계산 생물학자 아르빈드 라마나단(Arvind Ramanathan)은 “우리는 단백질 수준에서 유전자 수준 데이터로 이동하면 코로나 변이를 이해하기 위한 더 나은 모델을 구축하는 데 도움이 될 것이라고 가정했습니다. 전체 게놈과 진화에 나타나는 모든 변화를 추적하도록 모델을 훈련함으로써, 코로나뿐만 아니라 충분한 게놈 데이터가 있는 모든 질병에 대해 더 나은 예측을 할 수 있습니다”라고 말했습니다.
고성능 컴퓨팅의 노벨상으로 여겨지는 고든벨 상은 전 세계 100,000명에 이르는 컴퓨팅 전문가를 대표하는 컴퓨팅 기계 협회(ACM)가 주최하는 SC22 컨퍼런스에서 수여됩니다. 2020년부터 ACM은 HPC로 코로나에 대한 이해를 발전시킨 뛰어난 연구에 대해 특별상을 수여했습니다.
4글자 언어로 LLM 훈련
LLM은 일반적으로 수만 개의 단어로 배열되고, 더 긴 문장과 단락으로 결합될 수 있는 수십 개의 문자로 구성된 인간 언어에 대해 오랫동안 훈련됐습니다. 반면에 생물학 언어는 뉴클레오타이드를 나타내는 네 글자(DNA의 경우 A, T, G, C, RNA의 경우 A, U, G, C)만 서로 다른 유전자 염기서열로 배열되어 있죠.
글자 수가 적은 것이 AI에게는 더 간단한 과제로 보일 수 있지만, 생물학을 위한 언어 모델은 실제로 훨씬 더 복잡한데요. 인간의 경우 30억 개 이상의 뉴클레오타이드, 코로나바이러스의 경우 약 30,000개의 뉴클레오타이드로 구성된 게놈이 뚜렷하고 의미 있는 단위로 분해되기 어렵기 때문입니다.
라마나단은 “생명의 코드를 이해하는 데 있어 가장 큰 어려움은 게놈의 염기서열 분석 정보가 매우 방대하다는 것입니다. 뉴클레오타이드 서열의 의미는 인간의 텍스트에서 다음 문장이나 단락보다 훨씬 더 멀리 떨어져 있는 다른 서열의 영향을 받을 수 있습니다. 책의 몇 챕터에 해당하는 양을 넘을 수 있습니다”라고 말했습니다.
프로젝트에 참여한 NVIDIA 공동 연구자들은 LLM이 약 1,500개 뉴클레오타이드의 긴 문자열을 마치 문장인 것처럼 처리할 수 있도록 하는 계층적 확산 방법을 설계했습니다.
논문 공동 저자이자 NVIDIA의 AI 연구 수석 디렉터 겸 캘리포니아 공과대학교(Caltech)의 컴퓨팅 및 수학 과학 석좌 교수인 아니마 아난드쿠마르(Anima Anandkumar)는 “표준 언어 모델은 일관된 긴 염기서열을 생성하고 다양한 변이의 기본 분포를 학습하는 데 문제가 있습니다. 우리는 현실적인 변이를 생성하고 더 나은 통계를 수집할 수 있도록 더 높은 수준의 세부 사항에서 작동하는 확산 모델을 개발했습니다”라고 말했습니다.
코로나 주요 변이 바이러스 예측
연구팀은 먼저 세균 및 바이러스 생물정보학 자원센터(BV-BRC)의 오픈소스 데이터를 사용하여 박테리아와 같은 단세포 유기체인 원핵생물의 1억 1천만 개 이상의 유전자 서열에 대해 LLM을 사전 훈련했습니다. 그런 다음 코로나 바이러스에 대한 150만 개의 고품질 게놈 서열을 사용하여 모델을 미세 조정했습니다.
또한 연구원들은 더 광범위한 데이터 세트에 대한 사전 훈련을 통해 모델이 미래 프로젝트의 다른 예측 작업으로 일반화될 수 있도록 함으로써, 이러한 능력을 갖춘 최초의 전체 게놈 규모 모델 중 하나로 만들었습니다.
코로나 데이터에 대해 미세 조정된 LLM은 변이 바이러스의 게놈 서열을 구별할 수 있었습니다. 또한 과학자들이 미래의 주요 변이 바이러스를 예측하는 데 도움이 될 수 있는 코로나 게놈의 잠재적 변이를 예측하는 자체 뉴클레오타이드 서열을 생성할 수 있었습니다.
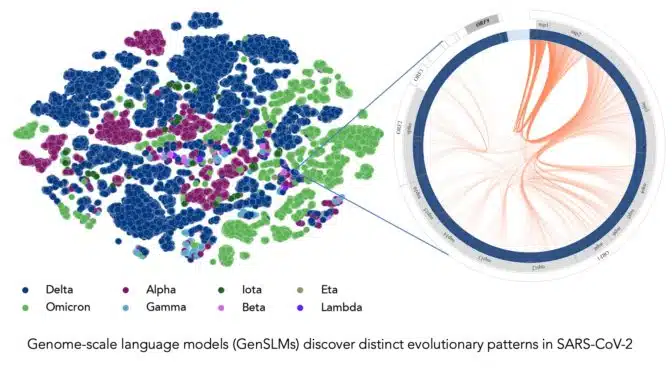
라마나단은 “대부분의 연구자들은 코로나 바이러스의 스파이크 단백질, 특히 인간 세포와 결합하는 도메인의 돌연변이를 추적해 왔습니다. 그러나 바이러스 게놈에는 빈번한 변이를 거치고 이해하는 데 중요한 다른 단백질이 있습니다”라고 말했습니다.
논문에 따르면, 이 모델은 또한 알파폴드(AlphaFold)와 오픈폴드(OpenFold)와 같이 인기 있는 단백질 구조 예측 모델과 통합되어, 연구자들이 바이러스 구조를 시뮬레이션하고 유전적 변이가 바이러스가 숙주를 감염시키는 능력에 미치는 영향을 연구하는 데 도움이 될 수 있다고 합니다. 오픈폴드는 LLM을 디지털 생물학과 화학 애플리케이션에 적용하는 개발자를 위해 NVIDIA BioNeMo LLM 서비스에 포함된 사전 훈련된 언어 모델 중 하나입니다.
GPU 가속 슈퍼컴퓨터로 AI 훈련 강화
연구팀은 아르곤의 폴라리스(Polaris), 미국 에너지부의 펄머터(Perlmutter), NVIDIA의 자체 Selene 시스템을 포함해 NVIDIA A100 Tensor Core GPU로 구동되는 슈퍼컴퓨터에서 AI 모델을 개발했습니다. 이러한 강력한 시스템으로 확장을 통해 1,500엑사플롭 이상의 훈련 실행 성능을 달성하여 현재 최대 규모의 생물학적 언어 모델을 생성하고 있습니다.
라마나단은 “현재 최대 250억 개의 파라미터가 있는 모델로 작업하고 있으며, 앞으로 이 수치는 크게 증가할 것으로 예상합니다. 모델 크기, 유전자 염기서열 길이, 필요한 훈련 데이터의 양은 수천 개의 GPU를 가진 슈퍼컴퓨터가 제공하는 계산 복잡성이 필수임을 의미합니다”라고 말했습니다.
연구원들은 25억 개의 파라미터가 있는 모델 버전을 약 4,000개의 GPU에서 훈련하는 데 한 달 이상이 소요된 것으로 추정합니다. 이미 생물학을 위한 LLM을 조사하고 있던 팀은 이번 논문과 코드를 공개 발표하기 전에 약 4개월간 프로젝트를 작업했는데요. 깃허브(GitHub) 페이지에는 폴라리스와 펄머터에서 모델을 실행하는 다른 연구원을 위한 지침이 포함되어 있습니다.
GPU 최적화 소프트웨어에 대해 NVIDIA NGC 허브에서 얼리 액세스로 사용할 수 있는 NVIDIA BioNeMo프레임워크는 여러 GPU에서 대규모 생체 분자 언어 모델을 확장하는 연구원들을 지원합니다. NVIDIA Clara Discovery의 신약 개발 도구 모음의 일부인 프레임워크는 화학, 단백질, DNA, RNA 데이터 형식을 지원합니다.
SC22에서 NVIDIA를 확인하고, 아래 특별 연설을 시청하세요.
상단의 이미지는 연구원의 LLM에 의해 염기서열 분석된 코로나 변종을 나타냅니다. 각 점은 코로나 변이에 따라 색상으로 구분됩니다. 이미지 제공: 아르곤 국립연구소의 바렛 케일, 맥스 즈비아긴, 마이클 E. 파프카.